
|
시장보고서
상품코드
2063869
의료기기 CRO 시장 : 점유율 분석, 업계 동향과 통계, 성장 예측(2026-2031년)Medical Device CRO - Market Share Analysis, Industry Trends & Statistics, Growth Forecasts (2026 - 2031) |
||||||
Mordor Intelligence에 의하면, 의료기기 CRO 시장 규모는 2025년 112억 1,000만 달러에서 2026년에는 119억 1,000만 달러로 확대되어 2031년까지 168억 2,000만 달러에 이를 것으로 예측되며, 2026년부터 2031년까지 CAGR 7.14%로 성장할 전망입니다.
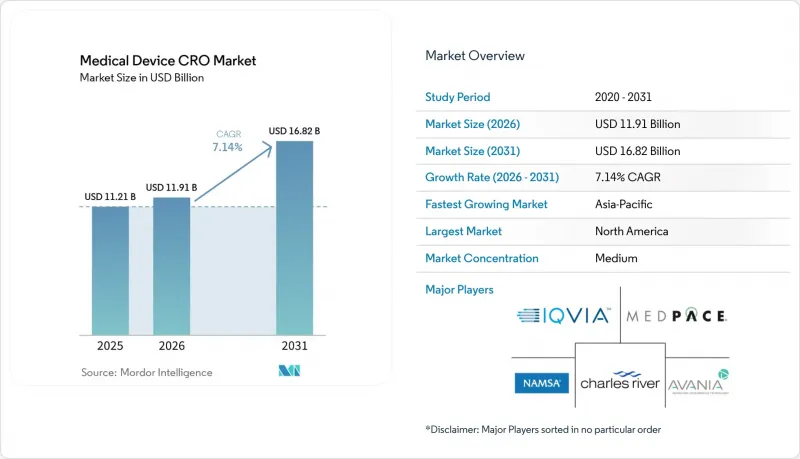
본 보고서는 사업 규모(전임상/시험, 임상), 서비스 유형(임상 모니터링, 임상시험 관리, 데이터 관리, 규제·메디컬 어페어즈, 메디컬 라이팅, 피험자 모집, 약물감시 등), 기기 분류(의료 기술, 진단, 휴대용 기기, 기타), 지역(북미, 유럽, 아시아태평양, 중동 및 아프리카, 남미)별로 분류되어 있습니다. 예상치(달러).
세계 의료기기 CRO 시장 동향과 인사이트
의료기기의 임상시험 규모와 복잡성이 커짐에 따라 아웃소싱이 활발해지고 있습니다.
의료기기 CRO 시장은 시판 전 승인을 훨씬 뛰어넘는 임상 증거 모델로부터 혜택을 받고 있습니다. 이는 시판 후 조사 및 PMCF가 일회성 활동이 아니라 지속적인 의무이기 때문입니다. MDCG 2025-10에 따르면, PMS는 의료기기의 수명 주기 전반에 걸쳐 적극적으로 데이터를 수집·분석해야 하며, 해당 정보는 위험 관리, 임상 평가, 기술 문서 및 시정 조치 프로세스에 반영되어야 한다고 규정되어 있습니다. 이러한 요건으로 인해, 특히 위험도가 높은 제품의 경우 체계적인 보고서 및 지속적인 업데이트가 요구되므로, 제조업체가 규제, 생물통계, 모니터링, 보고 등 모든 업무를 자체 팀 내에서 처리하기가 어려워지고 있습니다.
따라서 의료기기 CRO 시장은 신제품 출시뿐만 아니라 기존 제품과 관련된 지속적인 업무에서도 혜택을 보고 있습니다. 왜냐하면, 기존 의료기기가 새로운 규제 체계로 전환될 때 증거 부족 문제가 다시 대두되어, 새로운 아웃소싱 요청으로 이어질 가능성이 있기 때문입니다. 메드페이스(Medpace)는 2025 회계연도 매출이 전년 대비 20% 증가한 25억 3,020만 달러를 기록했다고 보고했으며, 2026 회계연도 매출은 27억 5,500만 달러에서 28억 5,500만 달러 사이가 될 것으로 전망하고 있습니다. 이는 개발 서비스 전반에 걸쳐 아웃소싱 수요가 견조한 추세를 보이고 있다는 견해를 뒷받침하는 것입니다.
EU의 MDR/IVDR에 따른 증거 및 PMCF 요건이 수요를 확대되고 있습니다.
MDCG 2025-10에 따라, PMS(시판 후 조사) 시스템은 품질 관리 시스템(QMS)의 필수적인 부분이 되었습니다. 이는 임상 증거 업무가 단순한 개별 신청 절차가 아니라, 지속적인 규정 준수 사이클의 일환으로 자리 잡게 되었음을 의미합니다. 또한, 본 지침에서는 PMCF 또는 PMPF 계획을 PMS 계획에 포함시켜야 하며, 그렇지 않을 경우 제조업체는 해당 활동이 적용되지 않는 이유를 정당화해야 한다고 규정하고 있습니다. PMCF 및 PMPF를 통해 얻은 인사이트는 임상 평가 또는 성능 평가, 유익성-위험성 평가, 라벨 표시, 기술 문서 및 기타 지속적인 규정 준수 기록을 갱신하는 데 활용되어야 합니다. 의료기기 CRO 시장에서 이러한 구조는 모니터링, 데이터 관리, 메디컬 라이팅, 비질런스 지원 및 규제 대응 업무에 대한 수요를 높이고 있습니다. 이는 스폰서가 승인 전뿐만 아니라 시판 후에도 근거 자료를 최신 상태로 유지해야 하기 때문입니다. MDR 및 IVDR 의무로 이미 손이 꽉 찬 사내 팀에서 스폰서가 이러한 업무의 상당 부분을 외부에 위탁하게 됨에 따라, 실세계 데이터(REW) 역량과 문서화 워크플로를 결합할 수 있는 공급업체는 더욱 유리한 입지를 차지하게 될 것입니다.
인증 기관의 역량 부족으로 인해 EU 인증 절차가 장기화되고 있습니다.
인증 기관의 역량 부족은 유럽 의료기기 CRO 시장에서 여전히 가장 뚜렷한 시기적 위험 요소 중 하나입니다. 이는 MDR 및 IVDR 절차를 거치는 업무량에 비해 인증 처리 속도가 여전히 따라가지 못하고 있기 때문입니다. MedTech Europe은 인증 기관을 규제상의 병목 현상으로 지적하고 있으며, 이러한 견해는 더욱 엄격해진 규정에 따라 승인, 갱신 및 증거 자료 갱신이 필요한 후원사들이 직면하고 있는 지속적인 압박과 일치합니다. 인증 시기가 늦어지면, 임상시험이나 문서 작성 작업이 이미 완료되었더라도 마일스톤 연계형 계약에 따라 CRO의 수익 인식이 지연될 가능성이 있습니다. 이는 의료기기 CRO 시장 수요를 감소시키지는 않겠지만, 유럽 내 납기 예측을 어렵게 만들며, 임상 업무와 규제 당국 간의 긴밀한 협력을 동시에 수행할 수 있는 제공업체의 가치를 높여줍니다. 실무적으로는 이러한 병목 현상으로 인해, 백로그의 위험을 관리하고 업무 흐름을 신중하게 조정하며, 장기간에 걸친 심사 기간 동안에도 후원사의 신뢰를 유지할 수 있는 기업으로 경쟁의 초점이 이동하고 있습니다.
부문별 분석
2025년 기준으로 임상 서비스는 의료기기 CRO 시장 점유율의 58.46%를 차지하고 있으며, 해당 부문은 2031년까지 연평균 성장률(CAGR) 7.67%로 성장할 것으로 전망됩니다. 이러한 선도적인 위상은 단순한 시험 건수 이상의 의미를 담고 있습니다. 왜냐하면 제조업체는 현재 시판 후 기간에도 계속해서 위험 관리 및 기술 문서 작성에 반영되어야 하는 임상적 의무를 지고 있기 때문입니다. MDCG 2025-10은 PMS(시판 후 조사)를 의료기기의 수명 주기 전반에 걸쳐 유익성·위험성 평가, 임상 평가, 시정 조치를 갱신해야 하는 능동적이고 지속적인 시스템으로 취급함으로써, 그 지속성을 명확히 하고 있습니다. 의료기기 CRO 시장에 있어 이는 임상 업무가 더 이상 최초 신청에만 집중되지 않게 되었음을 의미합니다. PMCF 및 관련 증거 자료를 유지·관리함으로써, 활동은 출시 후에도 장기간에 걸쳐 지속되기 때문입니다. 그 결과, 클래스 IIb, 클래스 III, 이식형 및 소프트웨어 의존도가 높은 제품을 취급하는 후원사의 경우, 사내에서 필요한 속도로 이러한 업무를 처리할 수 없을 때, 보다 대규모적이고 지속적인 아웃소싱이라는 선택지가 생겨나고 있습니다.
의료기기 CRO 시장에서 전임상 및 시험 서비스는 여전히 규모가 작지만, 이후의 많은 규제 및 임상적 결정의 업스트림 단계에 위치하기 때문에 전략적 중요성이 커지고 있습니다. 의료기기 후원사는 기존의 승인 당시 예상 사항이 아닌, 현재의 규제 요구 사항에 부합하는 생물학적 평가 계획, 화학적 특성 평가, 독성 평가 및 격차 분석을 여전히 필요로 하고 있습니다. 라보코프가 공개한 생체적합성 관련 서비스 내용을 살펴보면, 이러한 서비스가 이미 ISO 10993 시험, 화학적 특성 평가, 독성 위험 평가, 그리고 현행 규제 기준에 따른 기존 기기의 격차 분석 지원을 포괄하고 있음을 알 수 있습니다. 이로 인해 의료기기 CRO 업계는 개발 초기 단계에서 단순한 독립 시험 기관으로서 기능하는 것이 아니라, 실험실 업무와 허가 전략을 연계할 수 있는 시험 서비스 제공업체에 의존할 수밖에 없게 될 것입니다.
지역별 분석
2025년, 북미는 의료기기 CRO 시장 점유율의 45.34%를 차지하며 가장 큰 기여를 한 지역이 되었습니다. 이 분야의 상당 부분을 미국이 차지하고 있는데, 그 배경에는 특히 QMSR 시행 및 사이버 보안 요건 업데이트를 통해 규제 일정이 단기간에 여러 가지 준수 요건을 집중시킨 점이 있습니다. QMSR은 현재 ISO 13485:2016을 인용하여 반영하고 있으나, FDA 고유의 기록 관리 요건도 유지하고 있습니다. 즉, 제조업체는 ISO 인증만으로도 FDA의 감사나 문서화 의무가 대체된다고 가정하지 말고, 조화를 유지해야 합니다. 많은 스폰서들은 사내 품질 보증(QA) 및 규제 대응(RA) 담당자 채용에만 의존하지 않고, 아웃소싱을 통해 이러한 격차를 메우고 있으며, 이로 인해 지출이 억제되는 환경 속에서도 북미 의료기기 CRO 시장은 유지되고 있습니다. 이는 규제가 임상시험 건수뿐만 아니라 규정 준수 절차의 복잡화를 통해 아웃소싱 수요를 확대할 수 있음을 보여주는 가장 명확한 사례입니다.
유럽은 여전히 의료기기 CRO 시장에서 2위 지역 클러스터이며, 규제가 서비스 구성을 가장 직접적으로 재편하고 있는 지역이기도 합니다. MDR 및 IVDR에 따라 시판 후 증거 자료, 문서 관리, 인증 기관과의 소통이 업무 모델에 통합됨에 따라, 임상 업무와 규제 업무를 통합할 수 있는 CRO에 대한 의존도가 높아지고 있습니다. 동시에, 인증 기관의 병목 현상으로 인해 일정 관리가 계속해서 복잡해지고 있어, 계약 전환이나 마일스톤 달성 시기를 예측하기 어려워지는 반면, CRO에 대한 수요는 여전히 견조합니다. 따라서 유럽은 의료기기 CRO 시장에서 구조적인 수요 강세를 유지하고 있는 반면, 북미에 비해 수익 발생 시기와 관련된 리스크가 더욱 두드러지게 나타나는 지역입니다.
아시아태평양은 2031년까지의 의료기기 CRO 시장 규모에서 8.32%라는 가장 높은 연평균 성장률(CAGR)을 보일 것으로 전망됩니다. 해당 지역은 의료기기 개발 활동의 확대라는 혜택을 누리고 있지만, 현지 규제 절차의 특성상 현지에서의 대응 능력은 여전히 ‘선택 사항’이 아닌 ‘필수 사항’입니다. PMDA는 의료기기에 관한 공식적인 임상시험 신고 및 상담 체계를 유지하고 있으며, 이러한 절차들은 단일한 세계 템플릿이 아닌 현지화된 규제 대응의 필요성을 더욱 강조하고 있습니다. 이에 따라 일본 및 인근 시장에서 지역별 임상시험 기관에 대한 접근성, 언어 지원, 규제 당국 대응을 종합적으로 제공할 수 있는 서비스 제공업체에 대한 의료기기 CRO 시장 수요가 뒷받침되고 있습니다. 중국과 인도 역시 국내 제조 파이프라인이 임상 및 규제 관련 업무를 증가시키고, 후원사들이 아웃소싱을 선호하게 됨에 따라 미래 수요 기반을 확대되고 있습니다. 한국도 이 지역의 매력을 높이고 있습니다. 보다 효율적인 승인 환경이 하이브리드 시험 모델과 광범위한 지역에서의 시험 확대를 지원할 수 있기 때문입니다. 중동 및 아프리카 및 남미는 의료기기 CRO 시장에서 여전히 규모가 작지만, GCC 국가, 브라질, 아르헨티나에서 진행되는 다국적 등록 프로그램은 현지에서 사업을 전개할 수 있는 역량을 갖춘 국제적인 서비스 제공업체들에게 여전히 기회를 제공합니다.
기타 혜택:
- 엑셀 형식 시장 예측(ME) 시트
- 3개월간의 애널리스트 지원
자주 묻는 질문
목차
제1장 서론
제2장 조사 방법
제3장 주요 요약
제4장 시장 구도
제5장 시장 규모와 성장 예측
제6장 경쟁 구도
제7장 시장 기회와 향후 전망
JHS 26.06.23According to Mordor Intelligence, the medical device cRO market size is expected to increase from USD 11.21 billion in 2025 to USD 11.91 billion in 2026 and reach USD 16.82 billion by 2031, growing at a CAGR of 7.14% over 2026-2031.
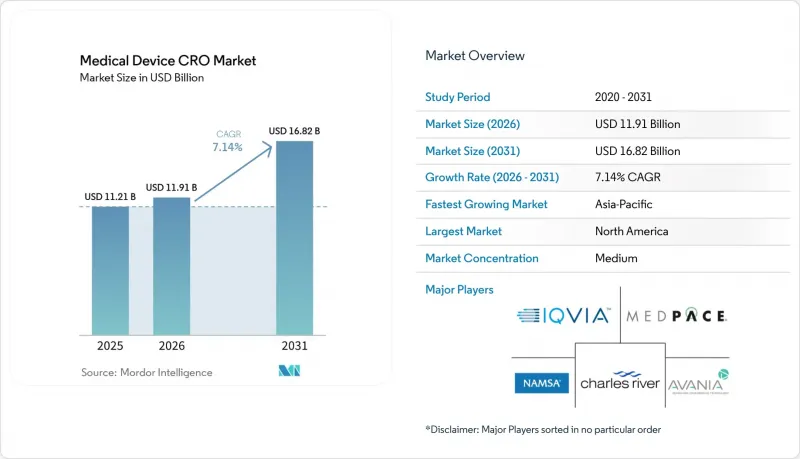
This report is Segmented by Scale of Operation (Preclinical/Testing, Clinical), Service Type (Clinical Monitoring, Trial Management, Data Management, Regulatory & Medical Affairs, Medical Writing, Patient Recruitment, Pharmacovigilance, and More), Device Class (MedTech, Diagnostic, Handheld, Others), and Geography (North America, Europe, Asia-Pacific, MEA, South America). Forecasts in Value (USD).
Global Medical Device CRO Market Trends and Insights
Outsourcing Intensifies as Device Trials Grow in Volume and Complexity
The medical device CRO market is gaining from a clinical evidence model that now stretches far beyond premarket submission, because post-market surveillance and PMCF are continuous obligations rather than one-time activities. MDCG 2025-10 states that PMS must actively gather and analyze data throughout the life of the device, and that this information must feed back into risk management, clinical evaluation, technical documentation, and corrective action processes. That requirement makes it harder for manufacturers to keep all regulatory, biostatistics, monitoring, and reporting tasks inside their own teams, especially when higher-risk products also require structured reports and ongoing updates.
The medical device CRO market therefore benefits from recurring work on legacy products as well as new launches, since older devices moving into newer frameworks can reopen evidence gaps and trigger fresh outsourcing mandates. Medpace reported full-year 2025 revenue of USD 2,530.2 million, up 20%, and guided for USD 2.755 billion to USD 2.855 billion in 2026, which supports the view that outsourcing demand remained healthy across the broader development services base.
EU MDR/IVDR Evidence and PMCF Requirements Expand Demand
MDCG 2025-10 makes the PMS system an integral part of the quality management system, which means clinical evidence work now sits inside a continuous compliance loop instead of a standalone filing exercise. The guidance also states that a PMCF or PMPF plan must be included within the PMS plan, or the manufacturer must justify why that activity is not applicable. Findings from PMCF and PMPF then have to update clinical or performance evaluation, benefit-risk assessment, labeling, technical documentation, and other ongoing compliance records. In the medical device CRO market, that structure increases demand for monitoring, data management, medical writing, vigilance support, and regulatory maintenance because sponsors must keep evidence current after launch and not only before approval. Providers that can combine real-world evidence capabilities with documentation workflows are better positioned as sponsors transfer more of this work outside internal teams that are already stretched by MDR and IVDR obligations.
Notified Body Capacity Bottlenecks Elongate EU Certifications
Notified Body capacity remains one of the clearest timing risks for the medical device CRO market in Europe because certification flow still lags the volume of work moving through MDR and IVDR pathways. MedTech Europe has described notified bodies as a regulatory bottleneck, and that framing aligns with the sustained pressure on sponsors that need approvals, renewals, and evidence updates under tighter rules. When certification timing slips, milestone-linked contracts can delay revenue recognition for CROs even if clinical or documentation work has already been completed. This does not remove demand from the medical device CRO market, but it does make European delivery schedules harder to predict and increases the value of providers that can combine clinical work with close regulatory coordination. In practical terms, the bottleneck shifts competition toward firms that can manage backlog risk, sequence workstreams carefully, and maintain sponsor confidence during long review cycles.
Other drivers and restraints analyzed in the detailed report include:
- FDA Cybersecurity Requirements and QMSR Alignment Elevate Software Validation and Cyber Testing Needs
- Decentralized Trial Adoption in Devices Increases DHT/eClinical and Remote-Operations Outsourcing
- DCT Patient Recruitment, Tech Access, and Data Governance Hurdles Slow Study Execution
For complete list of drivers and restraints, kindly check the Table Of Contents.
Segment Analysis
Clinical services held 58.46% of the medical device CRO market share in 2025, and the same segment is projected to grow at a 7.67% CAGR through 2031. This lead position reflects more than simple trial volume, because manufacturers now face clinical obligations that continue into the post-market period and feed back into risk management and technical documentation. MDCG 2025-10 makes that continuity explicit by treating PMS as an active, ongoing system that must update benefit-risk assessment, clinical evaluation, and corrective actions throughout the device lifetime. For the medical device CRO market, that means clinical work is no longer concentrated only around the first submission, since PMCF and related evidence maintenance extend activity well after launch. The result is a larger and more durable outsourcing pool for sponsors with Class IIb, Class III, implantable, and software-heavy products that cannot support these tasks internally at the required pace.
Preclinical and testing services remain smaller within the medical device CRO market, yet they are becoming more strategically important because they sit upstream of many later regulatory and clinical decisions. Device sponsors still need biological evaluation plans, chemical characterization, toxicological assessments, and gap analyses that align with current regulatory expectations rather than older approval assumptions. Labcorp's stated biocompatibility offering shows how these services already cover ISO 10993 testing, chemical characterization, toxicological risk assessment, and support for legacy-device gap reviews tied to current regulatory standards. This keeps the medical device CRO industry dependent on testing providers that can connect bench work to submission strategy, rather than acting only as isolated laboratories at the start of development.
Geography Analysis
North America held 45.34% of the medical device CRO market share in 2025, which made it the largest regional contributor. The United States accounts for most of this position because the regulatory calendar compressed several compliance demands into a short period, especially through QMSR implementation and updated cybersecurity expectations. QMSR now incorporates ISO 13485:2016 by reference, and it also keeps FDA-specific record controls in place, which means manufacturers must manage harmonization without assuming that ISO certification alone replaces FDA inspection or documentation duties. Many sponsors are filling this gap through outsourcing rather than by relying only on internal QA and RA hiring, which helps sustain the North American medical device CRO market even in a more disciplined spending environment. This makes the region the clearest example of how regulation can expand outsourcing demand through compliance complexity rather than through trial volume alone.
Europe remains the second-largest regional cluster in the medical device CRO market, and it is the geography where regulation most directly reshapes the service mix. MDR and IVDR have pushed more post-market evidence, documentation maintenance, and notified body interaction into the operating model, which increases dependence on CROs that can combine clinical and regulatory work. At the same time, notified body bottlenecks continue to complicate timing, which means CRO demand stays strong even while contract conversion and milestone pacing become harder to predict. This leaves Europe as a region with structural demand strength in the medical device CRO market, but also with more visible revenue-timing risk than North America.
Asia-Pacific is forecast to record the fastest 8.32% CAGR in the medical device CRO market size through 2031. The region benefits from expanding device development activity, but local regulatory pathways still make on-the-ground capability important rather than optional. PMDA maintains formal clinical trial notification and consultation structures for medical devices, and those processes reinforce the need for localized regulatory handling rather than a single global template. This supports demand in the medical device CRO market for providers that can combine regional site access, language support, and regulator-facing execution in Japan and neighboring markets. China and India are also broadening the future demand base as domestic manufacturing pipelines create more clinical and regulatory work that sponsors may prefer to outsource. South Korea adds to this regional appeal because a more efficient approval environment can support hybrid trial models and broader regional study footprints. Middle East and Africa, along with South America, remain smaller in the medical device CRO market, yet multinational registration programs in GCC countries, Brazil, and Argentina still create room for international providers with local operating reach.
- Avania
- BSI Group
- Charles River
- ClinChoice
- CROMSOURCE
- Emergo by UL
- Eurofins
- ICON
- Intertek
- IQVIA
- LabCorp
- MCRA
- MedPace
- NAMSA
- Nelson Labs
- Parexel International
- Premier Research
- ProPharma Group
- Qserve Group
- RQM+
- SGS
- Thermo Fisher Scientific
- TUV SUD
- Veranex
- WuXi App Tec
Additional Benefits:
- The market estimate (ME) sheet in Excel format
- 3 months of analyst support
TABLE OF CONTENTS
1 Introduction
- 1.1 Study Assumptions & Market Definition
- 1.2 Scope of the Study
2 Research Methodology
3 Executive Summary
4 Market Landscape
- 4.1 Market Overview
- 4.2 Market Drivers
- 4.2.1 Outsourcing Intensifies as Device Trials Grow in Volume and Complexity
- 4.2.2 EU MDR/IVDR Evidence and PMCF Requirements Expand Demand for Clinical, Regulatory, and Testing Services
- 4.2.3 FDA Cybersecurity Requirements and QMSR Alignment Elevate Software Validation and Cyber Testing Needs
- 4.2.4 Decentralized Trial Adoption in Devices Increases DHT/Eclinical and Remote-Operations Outsourcing
- 4.2.5 Cross-Border Data Transfer Constraints Drive Privacy Engineering and Data-Residency CRO Services
- 4.3 Market Restraints
- 4.3.1 Notified Body Capacity Bottlenecks Elongate EU Certifications and Add Planning Uncertainty
- 4.3.2 DCT Patient Recruitment, Tech Access, and Data Governance Hurdles Slow Study Execution
- 4.3.3 Cybersecurity SBOM/Patching Obligations Add Recurring QA/RA and Revalidation Burden
- 4.4 Value Chain Analysis
- 4.5 Regulatory Landscape
- 4.6 Technological Outlook
- 4.7 Porter's Five Forces
- 4.7.1 Threat of New Entrants
- 4.7.2 Bargaining Power of Suppliers
- 4.7.3 Bargaining Power of Buyers
- 4.7.4 Threat of Substitutes
- 4.7.5 Industry Rivalry
5 Market Size & Growth Forecasts (Value, USD)
- 5.1 By Scale of Operation
- 5.1.1 Preclinical/Testing
- 5.1.2 Clinical
- 5.2 By Service Type
- 5.2.1 Clinical Monitoring
- 5.2.2 Clinical Trial Management
- 5.2.3 Data Management & Biostatistics
- 5.2.4 Regulatory & Medical Affairs
- 5.2.5 Medical Writing
- 5.2.6 Patient & Site Recruitment
- 5.2.7 Safety & Pharmacovigilance
- 5.2.8 Others (DCT Enablement & eClinical Platforms, Imaging/Core Lab, etc.)
- 5.3 By Device Class
- 5.3.1 MedTech devices
- 5.3.2 Diagnostic devices
- 5.3.3 Handheld devices
- 5.3.4 Others
- 5.4 By Geography
- 5.4.1 North America
- 5.4.1.1 United States
- 5.4.1.2 Canada
- 5.4.1.3 Mexico
- 5.4.2 Europe
- 5.4.2.1 Germany
- 5.4.2.2 United Kingdom
- 5.4.2.3 France
- 5.4.2.4 Italy
- 5.4.2.5 Spain
- 5.4.2.6 Rest of Europe
- 5.4.3 Asia-Pacific
- 5.4.3.1 China
- 5.4.3.2 India
- 5.4.3.3 Japan
- 5.4.3.4 South Korea
- 5.4.3.5 Australia
- 5.4.3.6 Rest of Asia-Pacific
- 5.4.4 Middle East and Africa
- 5.4.4.1 GCC
- 5.4.4.2 South Africa
- 5.4.4.3 Rest of Middle East and Africa
- 5.4.5 South America
- 5.4.5.1 Brazil
- 5.4.5.2 Argentina
- 5.4.5.3 Rest of South America
- 5.4.1 North America
6 Competitive Landscape
- 6.1 Market Concentration
- 6.2 Market Share Analysis
- 6.3 Company Profiles {(includes Global level Overview, Market level overview, Core Segments, Financials as available, Strategic Information, Market Rank/Share for key companies, Products & Services, and Recent Developments)}
- 6.3.1 Avania
- 6.3.2 BSI Group
- 6.3.3 Charles River Laboratories
- 6.3.4 ClinChoice
- 6.3.5 CROMSOURCE
- 6.3.6 Emergo by UL
- 6.3.7 Eurofins Scientific
- 6.3.8 ICON plc
- 6.3.9 Intertek
- 6.3.10 IQVIA
- 6.3.11 Labcorp Drug Development
- 6.3.12 MCRA
- 6.3.13 Medpace
- 6.3.14 NAMSA
- 6.3.15 Nelson Labs
- 6.3.16 Parexel
- 6.3.17 Premier Research
- 6.3.18 ProPharma Group
- 6.3.19 Qserve Group
- 6.3.20 RQM+
- 6.3.21 SGS
- 6.3.22 Thermo Fisher Scientific
- 6.3.23 TUV SUD
- 6.3.24 Veranex
- 6.3.25 WuXi AppTec
7 Market Opportunities & Future Outlook
- 7.1 White-space & unmet-need assessment

